|
Twenty years ago, we isolated cDNA clones whose expressions were altered by pentylenetetrazole, a seizure-inducing drug, in murine brain. One of these genes, termed Sez12, is a mouse homolog of human DGCR2 gene that is located at the human chromosome 22q11.2 and that encodes a C-type lectin-like transmembrane protein. It is known that heterologous microdeletion in chromosome 22q11.2 causes a syndrome termed 22q11.2 deletion syndrome. 22q11.2 DS includes DiGeorge syndrome, conotruncal anomaly face syndrome, and velo-cardio-facial syndrome. At birth, congenital heart disease and immune disorder are typically seen in patients of 22q11.2 DS. Growth retardation including delayed motor development, facial dysmorphia, palatal anomalies, abnormal cerebellar morphology and early feeding problems are also common in 22q11.2 DS. Other characteristic signs and symptoms of 22q11.2 DS include psychiatric disorders with abnormal behavior, autistic spectrum disorders, and unprovoked seizures.
Individuals carrying the 22q11.2 microdeletion are also at risk for other psychiatric diseases, including anxiety and mood disorder. Recent evidence has indicated a relationship between the genes within 22q11.2 and clinical features of 22q11.2 DS, such as schizophrenia. Using mutant mice in our previous study, the deletion of gene for catechol-O-methyltransferase (Comt), rather than Dgcr2, seemed to be associated with an increased risk of schizophrenia in individuals of 22q11.2 DS. However, which of genes involved in the microdeletion of 22q11.2 DS are responsible for each clinical feature of 22q11.2 DS patients is remained unclear. Based on our previous findings including gene expression of Dgcr2, one of genes involved in the microdeletion of 22q11.2 DS, induced by a seizure-provoking reagent in cultured cells, we have hypothesized that loss of DGCR2 may revealed some neurological dysfunctions related to clinical manifestations of 22q11.2 DS that are caused by the complex genetic interactions.
|
|
 |
|
|
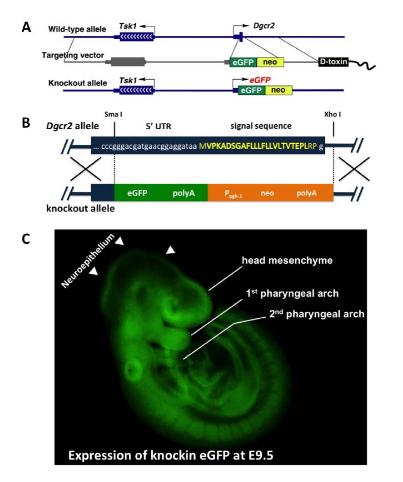
To investigate the functions of the Dgcr2 gene products, we have recently generated Dgcr2-knock-out/EGFP-knock-in (Dgcr2-KO) mice by gene targeting (A). In the targeting vector, a promotorless EGFP gene was inserted into the upstream of the initiation codon of Dgcr2 to visualize the pattern of Dgcr2 expression (B, C).
|
|
|
